Research projects
The discovery of stem cell-like cancer cells (cancer stem cells: CSCs) in human tumors opened the possibility for a new generation of anti-cancer therapies that target this critical population of cancer cells. CSCs are a subpopulation of cancer cells that are endowed with the defining characteristics of stem cells (self-renewal and multipotentiality) and the ability to initiate a tumor when transplanted. In theory, a single cancer stem cell can re-initiate tumor formation or form a metastatic tumor. Accumulating evidence indicates that CSCs are more resistant to chemo- and radiation-therapies compared to non-stem cancer cells from the same tumor, suggesting that targeting CSCs is necessary for improving long-term outcomes for cancer patients.
In an earlier study, we identified 45 genes that show differential expression patterns in glioma stem cells, compared to normal neural stem cells and non-stem glioma cells. Many of these genes are independent prognostic indicators of glioma patient survival and are expressed in a subset of cells in huma GBM tissues. We performed in depth analysis of one of these genes, S100A4, and validated that it is a novel marker and a regulator of glioma stem cells. We also showed that it is a master regulator of EMT and mesenchymal transition as it regulates expression of key EMT transcriptional regulators, such as SNAIL2 and ZEB1. Our findings suggest that inhibition S100A4 may simultaneously block glioma stem cell self-renewal/survival and mesenchymal transition in GBMs.
We are currently analyzing its mechanisms of action in promoting glioma stem cell survival and self-renewal and investigating its role in the crosstalk between glioma stem cell and tumor vasculature. In addition, we are developing inhibitors to block its function as potential new therapies for treating GBMs and other solid tumor.

Multi-omics analysis of brain cancer, integrating data from Single cell RNA sequencing, chip-seq, pathway analysis and patient survival data
Targeting regulators of immune suppression in brain cancer
One of the confounding factors for developing therapies that target CSCs is the heterogeneity of CSCs even within the same clinical tumor type. We discovered that a major contributor to CSC heterogeneity in the Ptch+/- model of medulloblastoma is the cell of origin. Using the CSC culture phenotype as an initial identifier of CSC subtypes, we identified three distinct tumor subtypes in the Ptch+/- model. These subtypes are histologically indistinguishable but molecularly distinct at the bulk tumor level and at the CSC level. We showed through cell type specific activation of the SHH pathway in vivo that transformation of neural stem cell and neural progenitor cells generate CSCs that are molecularly and cellularly distinct, particularly in their sensitivity to SHH inhibitors. Our work showed that CSC retain epigenetic memories of their cells of origin and that mitogenic pathways that drive CSC proliferation are the same as those driving its cell of origin.
We are currently testing multiple implications for understanding therapy resistance stemming from this study. First, this study demonstrated a novel mechanism of de novo therapy resistance to targeted therapy that does not require acquisition of new mutations but depends on the resident CSC phenotype. Second, it suggests that we may be able to predict a priori the types of mutations that will occur in therapy-resistant tumors: tumors in which CSCs depend on SHH signaling will necessarily acquire new mutations in the SHH pathway while tumors in which CSCs depend on other mitogenic pathways will not necessarily mutate within the SHH pathway. We are currently testing this hypothesis.
Elucidating cell: cell communication among different cell types in the brain tumor microenvironment
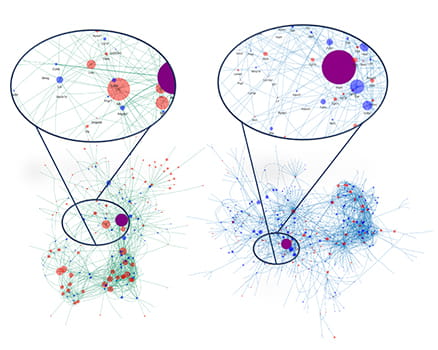
Single cell RNA sequencing from control and mutant medulloblastoma reveals differential cell: cell communication among various cell types in the tumor
Yap1 function in brain development and tumorigenesis
Developmental signaling pathways, such as the SHH, Notch, Wnt, and Hippo, are critical for stem cell regulation and cancer. We showed that Notch1 promotes NSC self-renewal in vivo in part by directly regulating the expression levels of limiting transcription factors in each of these pathways, including YAP1. YAP1 is a transcription effector of the Hippo pathway and is elevated in many human cancers, including brain cancer. We showed that YAP1 expression is sufficient to bypass the need for Notch pathway activation in NSC self-renewal in vitro. Our analysis of Yap1-/- brain indicates that YAP is also required for normal brain development. Currently, we are currently testing its role in brain tumor initiation and progression. Furthermore, we are studying its molecular mechanisms of function by identifying binding partners and downstream targets in normal NSCs and brain tumor stem cells. We are also developing novel small molecule inhibitors to block YAP1 function.
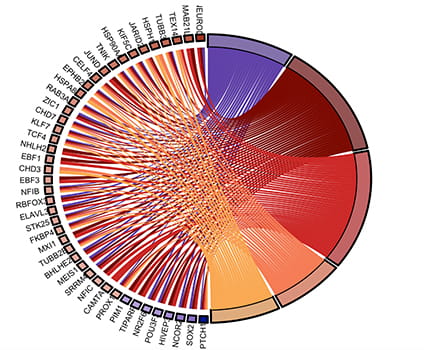
Yap1 promotes medulloblastoma stem cell maintenance while simultaneously suppressing their differentiation